


Fernández-Ramos
Research themes
Theoretical and Computational Chemistry: Simulation of chemical reactions
Main researcher(s)

Group members
| Le Thi Thanh, Hiep |
PhD Candidate |
|
| Lema Saavedra, Anxo |
PhD Candidate |
|
| Ferro Costas, David |
Inv. collaborator |
Research
Integrated Computational Protocol for Chemical Kinetics
The increasing accuracy of the electronic structure methods is allowing to tackle a great variety of chemical problems. In our research group we are contributing to the development of an integrated computational code able to simulate multiple aspects of chemical reactions dynamics. The methods developed can be applied to: organometallic catalysis, gas-phase mass spectrometry, the simulation of microwave spectra, the calculation of thermodynamic properties and to the study of many chemical reactions occurring in the gas phase. At the moment we are concentrating our efforts, although not exclusively, in applying the methods to:
- Combustion chemistry
- Chemistry at ultra-low temperatures
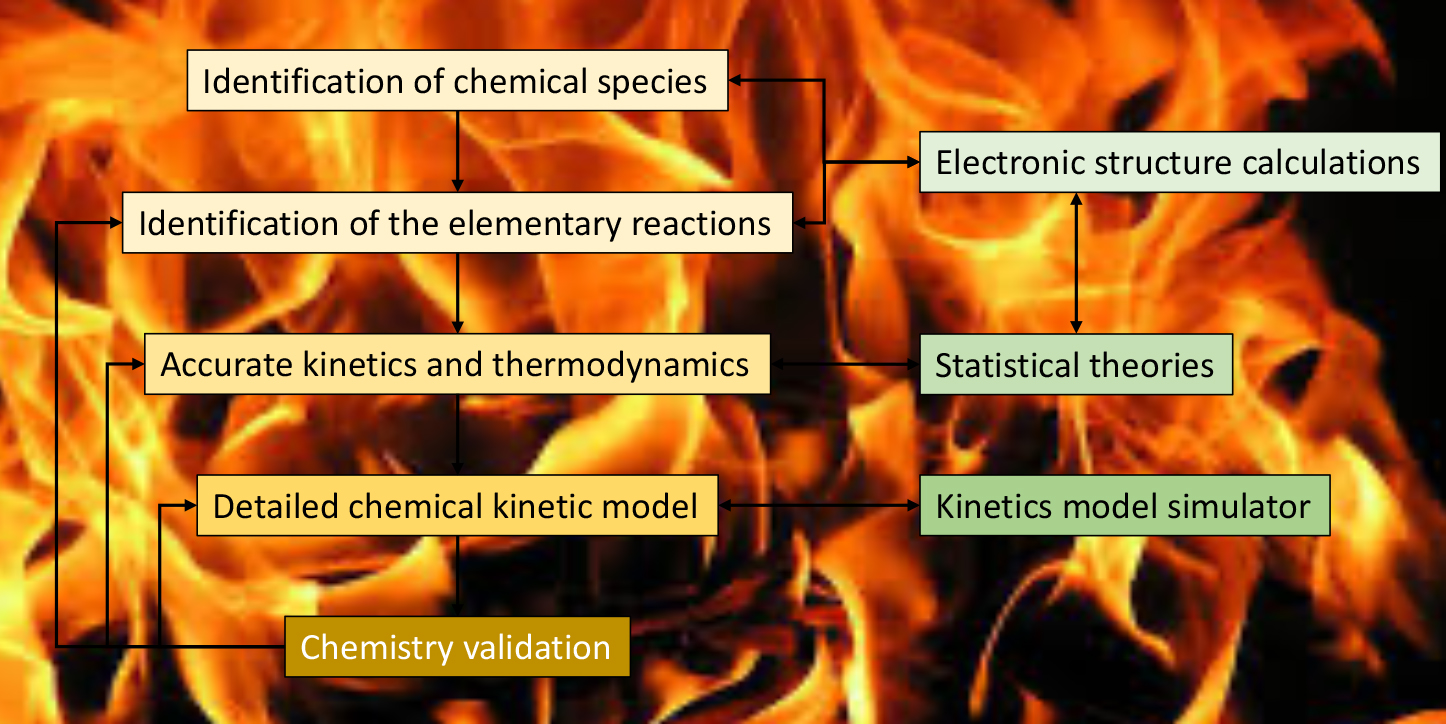 Combustion Chemistry
Combustion Chemistry
Emission reduction of noxious chemicals that are produced during the combustion of fossil fuels has become a world priority. One of the most promising substitutes are biofuels (fuels obtained from biomass). The study of the decomposition, oxidation, hydrogen abstraction and isomerization reactions of different biofuel candidates (for instance, alcohols, ethers and esters) is an important step to understand the combustion process as a whole.
However, the computer simulation of the combustion, which involves several hundreds of elementary reactions is a challenging task from several points of view. An example of how we handle this type of reactions is the decomposition reaction of the 1-propanol radicals (J. Phys. Chem. A, 2018, 122, 4790-4800). The flowchart below shows the process we adopted to design the theoretical kinetics mechanism for combustion:
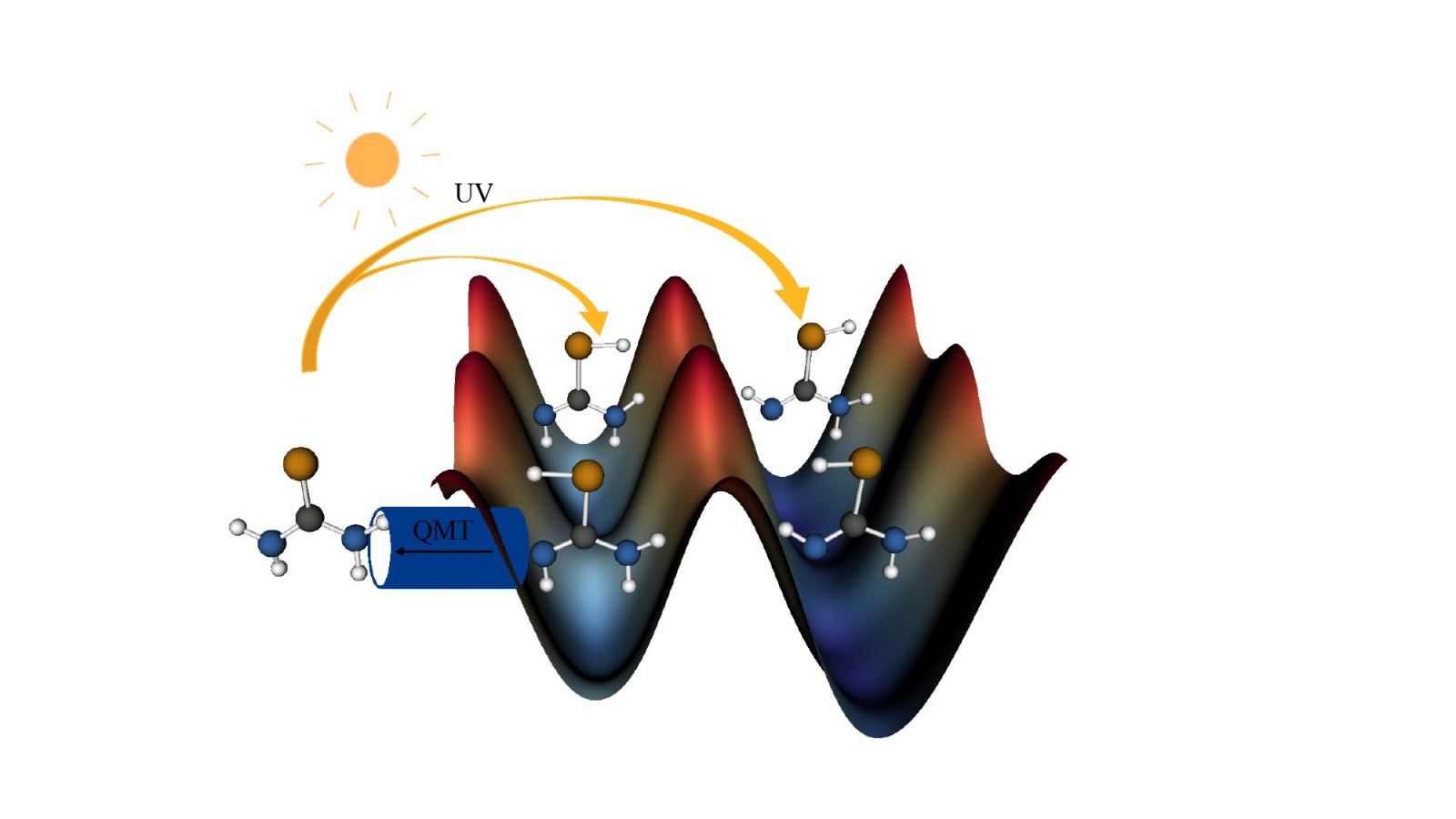
Chemistry at ultra-low temperatures
While in combustion reactions, factors as the conformational flexibility or torsional anharmonicity, play an important role in the reaction mechanism, at cryogenic temperatures the reaction processes are determined by quantum effects. In these conditions, reactions with an activation barrier can only occur by quantum mechanical tunneling (QMT), effect that allows particles to penetrate through classically forbidden regions of the potential barrier. The simulation of reactions occurring at very-low temperature require a fine tune of the potential energy surface, since the reaction occurs from the zero-point energy level of reactants. The combination of semiclassical methods to treat QMT and variational transition state theory (VTST) is able to mimic the reaction times of hydrogen transfer reactions. An example is the isomerization reaction that takes place in urea derivatives (selenourea and thiourea) in which the enol form is obtained by UV excitation and tautomerizes to the keto form (see figure below, where blue, brown, gray and white are the N, S, C and H atoms respectively). The reaction also involves subtle differences in stability between the enol conformers (Phys. Chem. Chem. Phys, 2020, 22, 24951-24963).
We are also studying bimolecular reactions in which participates a small radical (for instance, H or OH) and a small neutral molecule ("Kinetics of the Methanol Reaction with OH at Interstellar, Atmospheric, and Combustion Temperatures", J. Am. Chem. Soc., 2018, 140, 8, 2906-2918).
For some of these reactions (mainly the ones occurring with the radical OH) the thermal rate constants at ultra-low temperatures may be much larger than the ones at room temperature, which may be the reason for the formation of some of molecules in the interstellar space.
 Computer software
Computer software
Our research group has developed several software programs (all of them under MIT license) that form part of the Cathedral package. They can be downloaded freely from the following Github webpage.
- Pilgrim: A user-friendly program written in Python 3 (Comput. Phys. Commun., 2020, 256, 107457). It was designed to use direct-dynamics to calculate thermal rate constants of chemical reactions and to simulate chemical kinetics mechanisms. It was designed together with Donald G. Truhlar from the University of Minnesota (http://truhlar.chem.umn.edu).
For reaction processes with many elementary steps, each of these steps can be calculated using conventional transition state theory (TST) or VTST. In this version, Pilgrim can calculate thermal rate constants with the canonical version of the variational transition state theory (CVT), which requires the calculation of the minimum energy path (MEP) associated to each elementary step. Moreover, multi-dimensional quantum effects can be incorporated through the small-curvature tunneling approximation (SCT). The above methodologies are available for reactions involving a single structure and for reactions involving flexible molecules with multiple conformations. Specifically, for systems with many conformers the program can evaluate each of the elementary reactions by multi-path canonical variational transition state theory (MP-CVT) or multi-structural VTST (MS-VTST). Torsional anharmonicity can be also incorporated through the Torsiflex, MSTor or Q2DTor programs.
Once all the rate constants of the chemical processes of interest are known, by means of their calculation using Pilgrim or by using an analytical expression, it is possible to simulate the whole process using kinetic Monte Carlo (KMC). This algorithm allows performing a kinetics simulation and monitoring the evolution of each chemical species with time, as well as providing its chemical yield.
- Torsiflex: A program that searches for and locates all the conformational isomers of flexible acyclic molecules. It employs an efficient algorithm that combines systematic and stochastic search (Front. Chem. 2020, 8, 16).
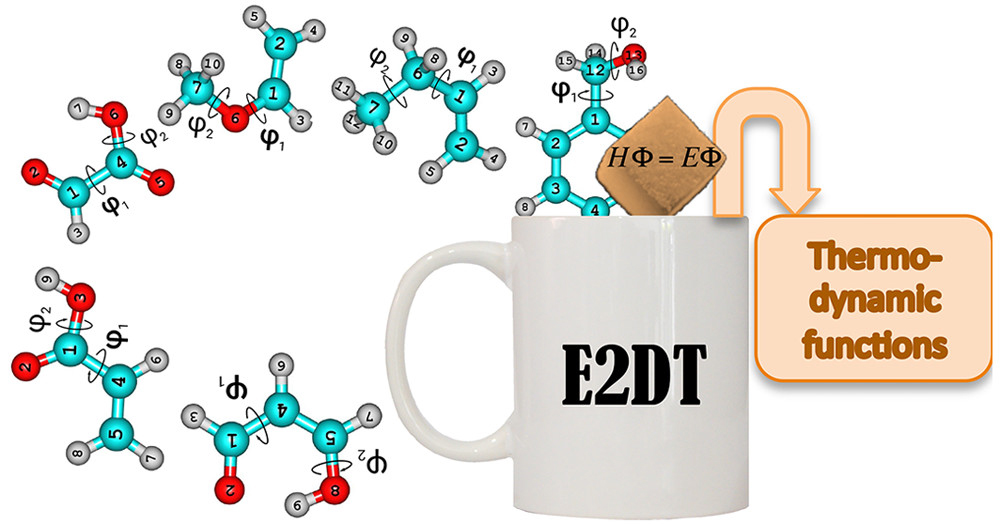 Q2DTor: A program to treat torsional anharmonicity trough pair torsions in flexible molecules. This software was designed to calculate accurate rovibrational partition functions and thermodynamic functions in flexible molecules using the extended tow-dimensional torsional (E2DT) method. The method can handle coupled pair torsions (Q2DTor: A program to treat torsional anharmonicity through coupled pair torsions in flexible molecules, Comput. Phys. Commun., 2018, 232, 190-205; Anharmonicity of Coupled Torsions, J. Chem. Theory Comput., 2017, 8, 3478-3492).
Q2DTor: A program to treat torsional anharmonicity trough pair torsions in flexible molecules. This software was designed to calculate accurate rovibrational partition functions and thermodynamic functions in flexible molecules using the extended tow-dimensional torsional (E2DT) method. The method can handle coupled pair torsions (Q2DTor: A program to treat torsional anharmonicity through coupled pair torsions in flexible molecules, Comput. Phys. Commun., 2018, 232, 190-205; Anharmonicity of Coupled Torsions, J. Chem. Theory Comput., 2017, 8, 3478-3492).
The software developed in our group can be linked to AutoMeKin, a program that employs graph theory to automate the search of transition states. The developer of AutoMeKin (E. Martinez-Núñez) is a member of the Theory and Computational Research Group of USC.